Hepatolenticular degeneration: report of three cases
DOI:
https://doi.org/10.17533/udea.iatreia.v30n4a07Keywords:
hepatolenticular degeneration, penicillamineAbstract
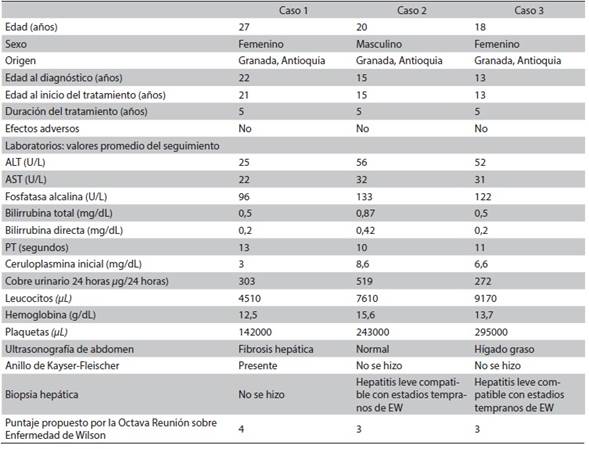
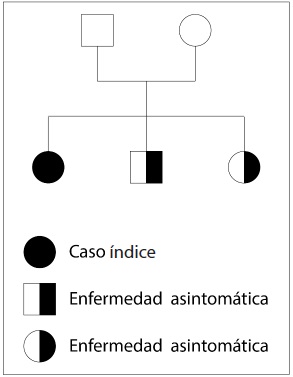
Hepatolenticular degeneration (Wilson disease) is a rare inherited disease that usually affects the liver, but may present in different forms and have multiple systemic complications. Diagnosis requires a high index of suspicion, mainly in young patients, and to take into account the main risk groups. Opportune and adequate treatment is important to avoid complications. We present three cases of this disease occurring in a family from Granada (Antioquia, Colombia), and treated at a III level institution in Cali (Colombia). The index case debuted with neuropsychiatric disorder, the second one was diagnosed on the basis of the family history and the third one started with steatohepatitis. The diagnostic score proposed by the Eighth Meeting on Wilson’s disease was 3 in two of the patients, and 4 in the third one. They were treated with D-penicillamine and monitored for 5 years, with minimal adverse events and no evidence of disease progression.
Downloads
References
(1.) Rodriguez-Castro KI, Hevia-Urrutia FJ, Sturniolo GC. Wilson’s disease: A review of what we have learned. World J Hepatol. 2015 Dec;7(29):2859-70. DOI 10.4254/wjh.v7.i29.2859.
(2.) Rodriguez-Castro KI, Hevia-Urrutia FJ, Sturniolo GC. Wilson’s disease: A review of what we have learned. World J Hepatol. 2015 Dec;7(29):2859-70. DOI 10.4254/wjh.v7.i29.2859.
(3.) Nanji MS, Nguyen VT, Kawasoe JH, Inui K, Endo F, Nakajima T, et al. Haplotype and mutation analysis in Japanese patients with Wilson disease. Am J Hum Genet. 1997 Jun;60(6):1423-9.
(4.) Beinhardt S, Leiss W, Stättermayer AF, Graziadei I, Zoller H, Stauber R, et al. Long-term outcomes of patients with Wilson disease in a large Austrian cohort. Clin Gastroenterol Hepatol. 2014 Apr;12(4):683-9. DOI 10.1016/j.cgh.2013.09.025.
(5.) European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012 Mar;56(3):671-85. DOI 10.1016/j.jhep.2011.11.007.
(6.) Diane W, Cox EAR. Wilson Disease. In: Feldman M, Friedman S, Lawrence JB. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. 10th ed. United States: Else vier; 2015. p. 1270-9.
(7.) Milkiewicz P, Saksena S, Hubscher SG, Elias E. Wilson’s disease with superimposed autoimmune features: report of two cases and review. J Gastroenterol Hepatol. 2000 May;15(5):570-4.
(8.) Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003 Jun;23(3):139-42.
(9.) Klein D, Lichtmannegger J, Heinzmann U, Summer KH. Dissolution of copper-rich granules in hepatic lysosomes by D-penicillamine prevents the development of fulminant hepatitis in Long-Evans cinnamon rats. J Hepatol. 2000 Feb;32(2):193-201.
(10.) Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson’s disease. N Engl J Med. 1987 Jul;317(4):209-13.
(11.) Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015 Jan;14(1):103-13. DOI 10.1016/S1474-4422(14)70190-5.

Published
How to Cite
Issue
Section
License
Copyright (c) 2017 Iatreia

This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Papers published in the journal are available for use under the Creative Commons license, specifically Attribution-NonCommercial-ShareAlike 4.0 International.
The papers must be unpublished and sent exclusively to the Journal Iatreia; the author uploading the contribution is required to submit two fully completed formats: article submission and authorship responsibility.