Sarcoidosis: muchas caras, una enfermedad. Revisión narrativa de la literatura
DOI:
https://doi.org/10.17533/udea.iatreia.11Palabras clave:
linfadenopatía, pulmón, sarcoidosisResumen
La sarcoidosis es una enfermedad granulomatosa sistémica de etiología desconocida. Esta puede afectar a pacientes de todas las latitudes y edades, siendo más frecuente entre la tercera y cuarta década de la vida con un segundo pico alrededor de los 50 años en las poblaciones escandinava y japonesa. Es más frecuente en mujeres y grave en la población afrodescendiente.
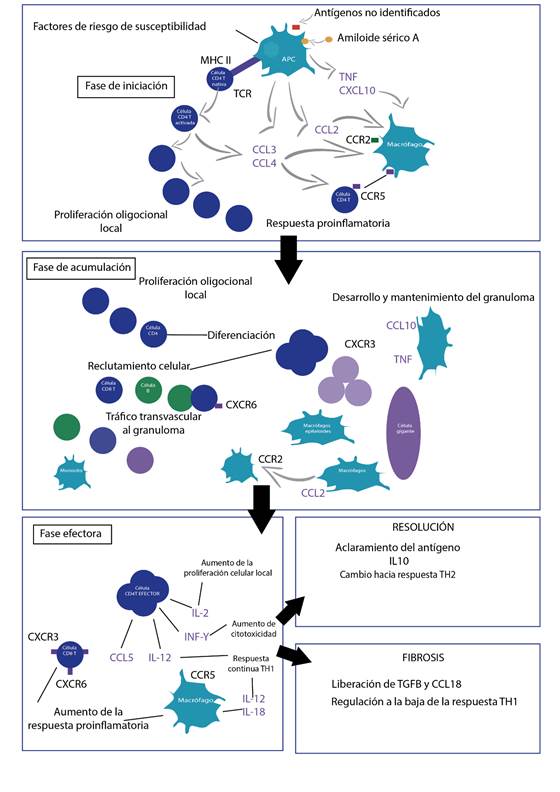
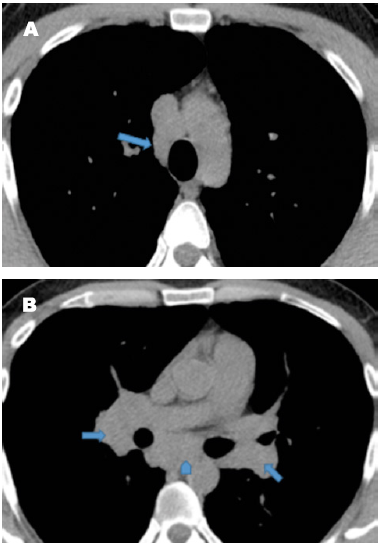
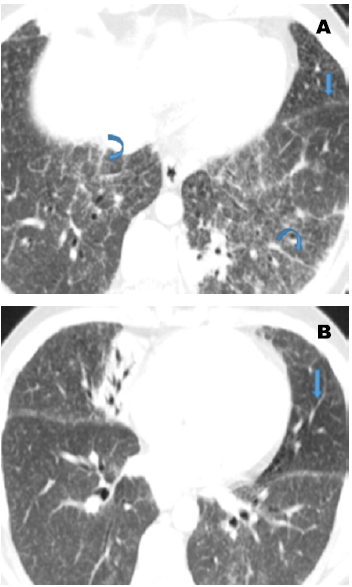
Los antígenos que inician esta respuesta granulomatosa son desconocidos, pero se presume que son aerotransportados por la alta frecuencia de compromiso pulmonar en esta enfermedad. Su presentación clínica abarca una amplia gama de manifestaciones, desde formas agudas y limitadas hasta el compromiso crónico con daño orgánico progresivo y muerte. Su diagnóstico se basa en la existencia de los granulomas no caseificantes en los tejidos, con la exclusión de otras enfermedades, entre ellas infección por micobacterias.
Descargas
Citas
(1.) Spagnolo P, Luppi F, Roversi P, Cerri S, Fabbri LM, Richeldi L. Sarcoidosis: challenging diagnostic aspects of an old disease. Am J Med. 2012 Feb;125(2):118-25. DOI 10.1016/j.amjmed.2011.06.003.
(2.) Johns CJ, Michele TM. The clinical management of sarcoidosis. A 50-year experience at the Johns Hopkins Hospital. Medicine (Baltimore). 1999 Mar;78(2):65-111.
(3.) Hem E. [Multiple benign sarcoid of the skin--100 years since Caesar Boeck's pioneering article]. Tidsskr Nor Laegeforen. 1999 Dec;119(30):4567-9.
(4.) Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011 Jan 26;305(4):391-9. DOI 10.1001/jama.2011.10.
(5.) Westney GE, Judson MA. Racial and ethnic disparities in sarcoidosis: from genetics to socioeconomics. Clin Chest Med. 2006 Sep;27(3):453-62, vi.
(6.) Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007 Nov;357(21):2153-65.
(7.) Lynch JP 3rd, Hwang J, Bradfield J, Fishbein M, Shivkumar K, Tung R. Cardiac involvement in sarcoidosis: evolving concepts in diagnosis and treatment. Semin Respir Crit Care Med. 2014 Jun;35(3):372-90. DOI 10.1055/s-0034-1376889.
(8.) Santamaria-Alza Y, Fajardo Rivero JE. Sarcoidosis: una serie de casos del nororiente colombiano. Neumol Cir Tórax. 2017 Ene-Mar;76(1):14-6.
(9.) Muñoz C, Restrepo-Escobar M, Martínez-Muñoz M, Echeverri A, Márquez J, Pinto LF. Differences Between Patients With Sarcoidosis With and Without Joint Involvement Treated for Fifteen Years in a Third Level Hospital. Reumatol Clin. 2018 Feb. Pii: S1699-258X(18)30021-4. DOI 10.1016/j.reuma.2018.01.001.
(10.) Judson MA, Baughman RP, Thompson BW, Teirstein AS, Terrin ML, Rossman MD, et al. Two year prognosis of sarcoidosis: the
ACCESS experience. Sarcoidosis Vasc Diffuse Lung Dis. 2003 Oct;20(3):204-11.
(11.) Co DO, Hogan LH, Il-Kim S, Sandor M. T cell contributions to the different phases of granuloma formation. Immunol Lett. 2004 Mar;92(1-2):135-42.
(12.) Song Z, Marzilli L, Greenlee BM, Chen ES, Silver RF, Askin FB, et al. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. 2005 Mar;201(5):755-67. Erratum in: J Exp Med. 2005 Sep 5;202(5):721.
(13.) Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H Jr, Bresnitz EA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001 Nov;164(10 Pt 1):1885-9.
(14.) Rybicki BA, Major M, Popovich J Jr, Maliarik MJ, Iannuzzi MC. Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol. 1997 Feb;145(3):234-41.
(15.) Carmona EM, Kalra S, Ryu JH. Pulmonary Sarcoidosis: Diagnosis and Treatment. Mayo Clin Proc. 2016 Jul;91(7):946-54. DOI 10.1016/j.mayocp.2016.03.004.
(16.) Hamzeh N. Sarcoidosis. Med Clin North Am. 2011 Nov;95(6):1223-34. DOI 10.1016/j.mcna.2011.08.004.
(17.) Chen ES, Moller DR. Etiology of sarcoidosis. Clin Chest Med. 2008 Sep;29(3):365-77, vii. DOI 10.1016/j.ccm.2008.03.011.
(18.) Haimovic A, Sanchez M, Judson MA, Prystowsky S. Sarcoidosis: a comprehensive review and update for the dermatologist: part I. Cutaneous disease. J Am Acad Dermatol. 2012 May;66(5):699.e1-18; quiz 717-8. DOI 10.1016/j.jaad.2011.11.965.
(19.) Govender P, Berman JS. The Diagnosis of Sarcoidosis. Clin Chest Med. 2015 Dec;36(4):585-602. DOI 10.1016/j.ccm.2015.08.003.
(20.) Chappity P, Kumar R, Sahoo AK. Heerfordt's Syndrome Presenting with Recurrent
Facial Nerve Palsy: Case report and 10-year literature review. Sultan Qaboos Univ Med J. 2015 Feb;15(1):e124-8.
(21.) Baughman RP. Pulmonary sarcoidosis. Clin Chest Med. 2004 Sep;25(3):521-30, vi.
(22.) Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999 Aug;160(2):736-55.
(23.) Judson MA. The Clinical Features of Sarcoidosis: A Comprehensive Review. Clin Rev Allergy Immunol. 2015 Aug;49(1):63-78. DOI 10.1007/s12016-014-8450-y.
(24.) Hillerdal G, Nöu E, Osterman K, Schmekel B. Sarcoidosis: epidemiology and prognosis. A 15-year European study. Am Rev Respir Dis. 1984 Jul;130(1):29-32.
(25.) Siltzbach LE, James DG, Neville E, Turiaf J, Battesti JP, Sharma OP, et al. Course and prognosis of sarcoidosis around the world. Am J Med. 1974 Dec;57(6):847-52.
(26.) Lynch JP 3rd, Kazerooni EA, Gay SE. Pulmonary sarcoidosis. Clin Chest Med. 1997 Dec;18(4):755-85.
(27.) Baughman RP, Winget DB, Bowen EH, Lower EE. Predicting respiratory failure in sarcoidosis patients. Sarcoidosis Vasc Diffuse Lung Dis. 1997 Sep;14(2):154-8.
(28.) Nishimura K, Itoh H, Kitaichi M, Nagai S, Izumi T. CT and pathological correlation of pulmonary sarcoidosis. Semin Ultrasound CT MR. 1995 Oct;16(5):361-70.
(29.) Brauner MW, Grenier P, Mompoint D, Lenoir S, de Crémoux H. Pulmonary sarcoidosis: evaluation with high-resolution CT. Radiology. 1989 Aug;172(2):467-71.
(30.) Müller NL, Miller RR. Ground-glass attenuation, nodules, alveolitis, and sarcoid granulomas. Radiology. 1993 Oct;189(1):31-2.
(31.) Hamper UM, Fishman EK, Khouri NF, Johns CJ, Wang KP, Siegelman SS. Typical and atypical CT manifestations of pulmonary sarcoidosis. J Comput Assist Tomogr. 1986 Nov-Dec;10(6):928-36.
(32.) Reich JM. Mortality of intrathoracic sarcoidosis in referral vs population-based settings: influence of stage, ethnicity, and corticosteroid therapy. Chest. 2002 Jan;121(1):32-9.
(33.) Davies CW, Tasker AD, Padley SP, Davies RJ, Gleeson FV. Air trapping in sarcoidosis on computed tomography: correlation with lung function. Clin Radiol. 2000 Mar;55(3):217-21.
(34.) Bartz RR, Stern EJ. Airways obstruction in patients with sarcoidosis: expiratory CT scan findings. J Thorac Imaging. 2000 Oct;15(4):285-9.
(35.) Abehsera M, Valeyre D, Grenier P, Jaillet H, Battesti JP, Brauner MW. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. AJR Am J Roentgenol. 2000 Jun;174(6):1751-7.
(36.) Jamilloux Y, Kodjikian L, Broussolle C, Sève P. Sarcoidosis and uveitis. Autoimmun Rev. 2014 Aug;13(8):840-9. DOI 10.1016/j.autrev.2014.04.001.
(37.) Martínez-Berriotxoa A, Fonollosa A, Artaraz J. [Uveitis: diagnostic approach]. Rev Clin Esp. 2012 Oct;212(9):442-52. DOI 10.1016/j.rce.2011.12.004.
(38.) Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am. 2013 May;39(2):277-97. DOI 10.1016/j.rdc.2013.02.007.
(39.) Kansal V, Dollin M. Ocular involvement in sarcoidosis. CMAJ. 2017 Apr;189(16):E609. DOI 10.1503/cmaj.160569.
(40.) Zapata-González F, Vásquez-Ochoa LA, Arroyave-Sierra JE, Arredondo-Ossa MI, Molina-Vélez V, Río-Cobaleda D. Sarcoidosis cutánea. CES Med. 2014 Jul- Dic;28(2):293-306.
(41.) Valovis R. Sarcoidosis estudio clínico de 51 casos y revisión de la literatura. Acta Médica Colomb. 1977;2(2):101-10.
(42.) Visser H, Vos K, Zanelli E, Verduyn W, Schreuder GM, Speyer I, et al. Sarcoid arthritis: clinical characteristics, diagnostic aspects, and risk factors. Ann Rheum Dis. 2002 Jun;61(6):499-504.
(43.) Banse C, Goëb V. Do not forget the joint involvement of sarcoidosis. Immunotherapy. 2015;7(6):599-600. DOI 10.2217/imt.15.24.
(44.) Kidd DP. Sarcoidosis of the central nervous system: clinical features, imaging, and CSF results. J Neurol. 2018 Aug;265(8):1906-15. DOI 10.1007/s00415-018-8928-2.
(45.) Rocha ÓG, García PK, Echeverri JE, D’Achiardi RE, Rodríguez MP, Córdoba JP, et al. Sarcoidosis y compromiso renal: reporte de un caso y revisión de la literatura científica. Univ Méd. 2012 Ene-Mar;53(1):94-106.
(46.) Holmes J, Lazarus A. Sarcoidosis: extrathoracic manifestations. Dis Mon. 2009 Nov;55(11):675-92. DOI 10.1016/j.disamonth.2009.05.002.
(47.) Okumus G, Musellim B, Cetinkaya E, Turker H, Uzaslan E, Yenturk E, et al. Extrapulmonary involvement in patients with sarcoidosis in Turkey. Respirology. 2011 Apr;16(3):446-50. DOI 10.1111/j.1440-1843.2010.01878.x.
(48.) Valeyre D, Bernaudin JF, Uzunhan Y, Kambouchner M, Brillet PY, Soussan M, et al. Clinical presentation of sarcoidosis and diagnostic work-up. Semin Respir Crit Care Med. 2014 Jun;35(3):336-51. DOI 10.1055/s-0034-1381229.
(49.) Bonfioli AA, Orefice F. Sarcoidosis. Semin Ophthalmol. 2005 Jul-Sep;20(3):177-82.
(50.) Harvey J, Catoggio L, Gallagher PJ, Maddison PJ. Salivary gland biopsy in sarcoidosis. Sarcoidosis. 1989 Mar;6(1):47-50.
(51.) Blaise P, Fardeau C, Chapelon C, Bodaghi B, Le Hoang P. Minor salivary gland biopsy in diagnosing ocular sarcoidosis. Br J Ophthalmol. 2011 Dec;95(12):1731-4. DOI 10.1136/bjophthalmol-2011-300129.
(52.) Truedson H, Stjernberg N, Thunell M. Scalene lymph node biopsy. A diagnostic method in sarcoidosis. Acta Chir Scand. 1985;151(2):121-3.
(53.) Karagiannidis A, Karavalaki M, Koulaouzidis A. Hepatic sarcoidosis. Ann Hepatol. 2006 Oct-Dec;5(4):251-6.
(54.) Ungprasert P, Carmona EM, Crowson CS, Matteson EL. Diagnostic Utility of Angiotensin-Converting Enzyme in Sarcoidosis: A Population-Based Study. Lung. 2016 Feb;194(1):91-5. DOI 10.1007/s00408-015-9826-3.
(55.) Lieberman J. Elevation of serum angiotensin-converting-enzyme (ACE) level in sarcoidosis. Am J Med. 1975 Sep;59(3):365-72.
(56.) Stouten K, van de Werken M, Tchetverikov I, Saboerali M, Vermeer HJ, Castel R, et al. Extreme elevation of serum angiotensin-converting enzyme (ACE) activity: always consider familial ACE hyperactivity. Ann Clin Biochem. 2014 Mar;51(Pt 2):289-93. DOI 10.1177/0004563213489812.
(57.) Studdy PR, Bird R. Serum angiotensin converting enzyme in sarcoidosis—its value in present clinical practice. Ann Clin Biochem. 1989 Jan;26 (Pt 1):13-8.
(58.) Gupta D, Chetty M, Kumar N, Aggarwal AN, Jindal SK. Anergy to tuberculin in sarcoidosis is not influenced by high prevalence of tuberculin sensitivity in the population. Sarcoidosis Vasc Diffuse Lung Dis. 2003 Mar;20(1):40-5.
(59.) Siltzbach LE. The Kveim test in sarcoidosis. A study of 750 patients. JAMA. 1961 Nov;178:476-82.
(60.) Sulavik SB, Spencer RP, Palestro CJ, Swyer AJ, Teirstein AS, Goldsmith SJ. Specificity and sensitivity of distinctive chest radiographic and/or 67Ga images in the noninvasive diagnosis of sarcoidosis. Chest. 1993 Feb;103(2):403-9.
(61.) Judson MA, Costabel U, Drent M, Wells A, Maier L, Koth L, et al. The WASOG Sarcoidosis Organ Assessment Instrument: An update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis. 2014 Apr;31(1):19-27.
(62.) Bickett AN, Lower EE, Baughman RP. Sarcoidosis Diagnostic Score: A Systematic Evaluation to Enhance the Diagnosis of Sarcoidosis. Chest. 2018 Nov;154(5):1052-60. DOI 10.1016/j.chest.2018.05.003.
(63.) Newman LS, Rose CS, Maier LA. Sarcoidosis. N Engl J Med. 1997 Apr;336(17):1224-34. Erratum in: N Engl J Med 1997 Jul;337(2):139.
(64.) Judson MA. The diagnosis of sarcoidosis. Clin Chest Med. 2008 Sep;29(3):415-27, viii. DOI 10.1016/j.ccm.2008.03.009.
(65.) Verbsky JW, Routes JM. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med. 2014 Jun;35(3):330-5. DOI 10.1055/s-0034-1376862.
(66.) Ramos-Casals M, Brito-Zerón P, García-Carrasco M, Font J. Sarcoidosis or Sjögren syndrome? Clues to defining mimicry or coexistence in 59 cases. Medicine (Baltimore). 2004 Mar;83(2):85-95.
(67.) Yu JQ, Doss M, Codreanu I, Zhuang H. PET/CT in Patients with Sarcoidosis or IgG4 Disease. PET Clin. 2012 Apr;7(2):191-210. DOI 10.1016/j.cpet.2012.01.005.
(68.) Yu J, Hall T, Kale S, Phillips A, Madge S. IgG4 disease: a revised diagnosis of sarcoidosis after 36 years of treatment. Br J Hosp Med (Lond). 2013 Aug;74(8):470-1.
(69.) Michel L, Clairand R, Néel A, Masseau A, Frampas E, Hamidou M. Association of IgG4-related disease and sarcoidosis. Thorax. 2011 Oct;66(10):920-1. DOI 10.1136/thx.2011.160341.
(70.) Ishii S, Miyajima M, Sakuma K, Kikuchi K, Shishido F. Comparison between sarcoidosis and IgG4-related disease by whole-body 67Ga scintigraphy. Nucl Med Commun. 2013 Jan;34(1):13-8.DOI 10.1097/MNM.0b013e32835a2eea.
(71.) Baughman RP, Grutters JC. New treatment strategies for pulmonary sarcoidosis: antimetabolites, biological drugs, and other treatment approaches. Lancet Respir Med. 2015 Oct;3(10):813-22. DOI 10.1016/S2213-2600(15)00199-X.

Descargas
Publicado
Cómo citar
Número
Sección
Licencia
Derechos de autor 2019 Iatreia

Esta obra está bajo una licencia internacional Creative Commons Atribución-CompartirIgual 4.0.
Los artículos publicados en la revista están disponibles para ser utilizados bajo la licencia Creative Commons, específicamente son de Reconocimiento-NoComercial-CompartirIgual 4.0 Internacional.
Los trabajos enviados deben ser inéditos y suministrados exclusivamente a la Revista; se exige al autor que envía sus contribuciones presentar los formatos: presentación de artículo y responsabilidad de autoría completamente diligenciados.